Research Topics
Explore the key research themes, active projects, and scientific investigations carried out at the Renjith Thomas Lab.
Software we use are listed (incomplete list)
Quantum Chemistry: Gaussian 09W (licence), Orca (open access)
Turbomole (licence)
Wavefunction: QTAIM (licence), Multiwfn
Conformations: GFNn-xTB (n=0,1,2), GFN-FF, and g-xTB (through atomistica.online), Vconf
Visualisation: Gaussview 5.0, Chemcraft (licence), Avogadro (open access)
Docking: Autodock Vina4 (open acess)
Hardware: Xeon 20 core with 128 GB RAM, Xeon 16 core with 64 GB RAM, I9 20 core with 64 GB RAM, I7 10 core with 32 GB RAM
Cluster access through BRAF, PARAM Utkarsh (CDAC), ACCESS (harware and software)
Data Ananlysis and ML: Jamovi (open access) and Python
Berchmans Protocol for Modelling Non-Covalent Interactions 1.0 (BerchNCI 1.0)
Please read our article at https://publishing.aidasco.org/journals/index.php/aire/article/view/84/152
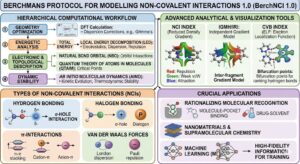
The development of the BerchNCI 1.0 protocol marks a significant advancement in our ability to decode the “electronic anatomy” of the subtle forces that govern molecular world. While individually weak, these non-covalent interactions (NCIs) ranging from archetypal hydrogen bonds to sophisticated -hole and -system interactions are the fundamental organizing principles behind protein folding, DNA stability, and the efficacy of modern drug design. The BerchNCI 1.0 protocol systematizes the study of these forces by integrating high-level geometry optimization with a hierarchical suite of analytical tools, including the Non-Covalent Interaction (NCI) index, Quantum Theory of Atoms in Molecules (QTAIM), and Natural Bond Orbital (NBO) analysis. By moving beyond simple static observations, the protocol utilizes Ab Initio Molecular Dynamics (AIMD) to capture the temporal evolution and thermodynamic stability of molecular assemblies, providing a “molecular movie” of interactions as they occur in real-time.
The importance of this standardized methodology lies in its power to resolve competitive interactions and rationalize molecular recognition with benchmark precision. By employing Local Energy Decomposition (LED), BerchNCI 1.0 dissects the total interaction energy into its physical drivers such as electrostatics, dispersion, and Pauli repulsion offering researchers a clear, quantitative understanding of why a specific molecular complex is stable. This comprehensive approach does more than just visualize the “invisible” world of weak chemical bonds; it generates the high-fidelity data essential for training future machine learning models to predict molecular properties. As a computational framework, BerchNCI 1.0 serves as an indispensable partner to experimental techniques, bridging the gap between fundamental molecular theory and practical innovation in fields as diverse as pharmacology, materials science, and sustainable catalysis.
Modeling of Polymer-Solvent Systems & Blends
- Dr Athira Maria John (Post Doc)
- Dr Rehin Sulay (Post Doc)

We also work on the molecular modeling of polymer-solvent systems and polymer blends, specifically targeting the electronic-level interactions that dictate material performance and environmental fate. By employing a multilevel computational approach combining DFT with the topological tools such as QTAIM, NCI, and NBO we decode the complex solvation blueprint of polymers like PLA and PEG. For instance, our work on PLA reveals that its aqueous stability is a delicate balance between directional H-bonds at carbonyl sites and internal steric repulsion, while AIMD simulations provide a real-time view of the proton transfer and ester bond cleavage that trigger hydrolytic degradation. This high-level theoretical framework allows for the rational design of next-generation green plastics with precisely tuned degradation kinetics, moving beyond traditional trial-and-error methods.
In parallel, we investigate the structural stability and solubility of PEG and MPEG systems to enhance their functionality in pharmaceutical and industrial applications. By generating MEP maps and utilizing IGMH analysis, we identify the specific ether oxygen sites where water molecules anchor, confirming that these weak yet vital non-covalent interactions are the electronic origin of polymer biocompatibility. Through molecular docking, we also bridge the gap between chemistry and biology by identifying how microbial enzymes bind to these polymer chains with high affinity. Collectively, our research provides a mechanistic basis for predicting how polymer-solvent interactions and blend morphologies influence everything from solubility to long-term breakdown, offering a transformative toolkit for developing sustainable, high-performance materials for environmental and biomedical interests.
Aromaticity and Proton Sponges
- Manjesh Mathew (PhD student)
- Prince Sebastian (MSc student)
M. Mathew, R. Puchta, R. Thomas, Proton affinity Revisited: Benchmarking computational approaches for accurate predictions, Comput. Theor. Chem. 1233 (2024) 114477. https://doi.org/10.1016/j.comptc.2024.114477
M. Mathew, R. Puchta, R. Thomas, Protonation-Induced Planarization and Aromaticity Enhancement in Diazahelicenes: A Route to Stronger Bases, J. Phys. Chem. A. (2026). https://doi.org/10.1021/ACS.JPCA.5C06393.
The study of aromaticity has evolved from a static structural concept into a versatile tool for moderating the electronic behavior of complex organic systems. Recent investigations into diazahelicenes have demonstrated that these polycyclic aromatic compounds can function as potent “proton sponges,” with their exceptional basicity deeply rooted in protonation-induced structural changes. Computational analyses using DFT methods, specifically the M06-2X functional, reveal that protonation minimizes the repulsion between closely spaced nitrogen atoms, driving the molecules toward greater planarity. This geometric shift, confirmed by HOMA and NICS indices, enhances electron delocalization and aromaticity, providing a fundamental mechanistic link between the “frustrated” neutral state of heterohelicenes and their high basic strength. These findings not only expand the understanding of structure-property relationships in helicene chemistry but also provide a rational basis for designing new superbases tailored for applications in catalysis and molecular recognition. This study has been conducted in close collabortation with Dr Ralph Puchta, University of Earlangen, Germany.
Beyond internal structural modifications, aromaticity can be systematically tuned through the strategic use of binary solvent mixtures. Research on nitroaniline isomers shows that π-electron delocalization is not an intrinsic constant but responds strongly to solvent polarity and hydrogen-bond strength. While moving from the gas phase to a polar solution typically leads to a loss of aromatic stabilization, the introduction of a less polar co-solvent can moderate these effects, restoring delocalization indices by significant margins. Isomer-dependent behavior is a critical factor in this modulation; the para isomer exhibits the highest sensitivity to environmental perturbations due to strong donor-acceptor communication through the ring, whereas the meta isomer remains relatively stable. By correlating AIM-based hydrogen bond energies with magnetic and geometric aromaticity criteria, this work establishes solvent mixing as a practical, molecular-level tool for controlling charge-transfer behavior and electronic structure without altering the underlying molecular framework.
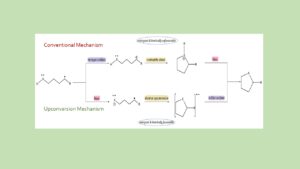
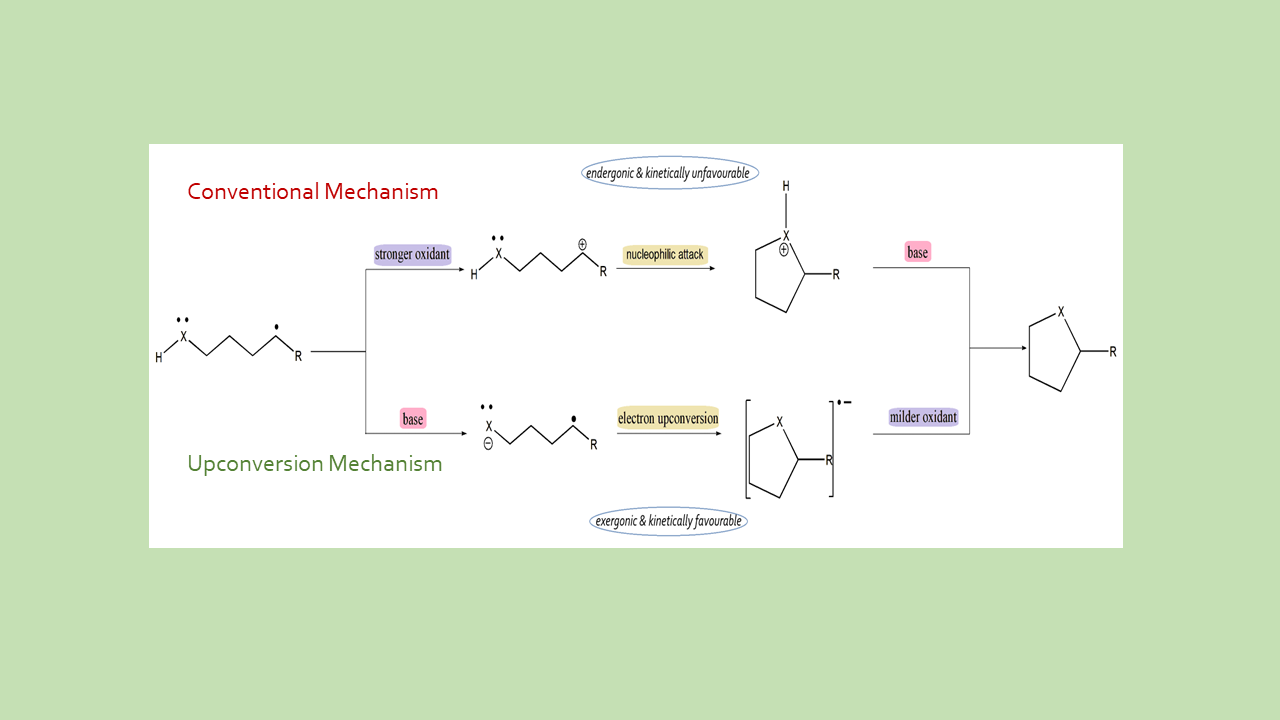
Electron Upconversion (EUC) in molecular catalysis
- Meera Kattoor PhD Student
With active support of our collaborator Prof Igor Alabugin from Florida State University, USA , we have examined the fundamental understanding of electronic structures to drive practical innovation, as exemplified by our work on Electron Upconversion (EUC) in molecular catalysis. We establishes the EUC as a computational framework for characterizing catalysts that drive high-energy chemical transformations through electronic excitation. Traditional energy profiles are insufficient for a complete understanding of catalytic efficiency; instead, the EUC protocol successfully maps the electronic anatomy of a reaction by integrating IGMH and NBO analysis to resolve hidden electronic rearrangements and donor-acceptor interactions. A key result is the protocol’s ability to precisely quantify the contributions of individual atomic fragments to the stabilization of transient states, providing a standardized, predictive methodology for the rational design of light-harvesting systems. These high-fidelity electronic descriptors provide the essential data foundation required to train future machine learning models for the autonomous discovery of sustainable catalysts.
Please see our paper in the Journal of Computational Chemistry – Electron Upconversion Enables C-P and C-S Bond Formation Under Mild Oxidative Conditions: A Theoretical Study
Computational Study of Catalytic Reactions
- Dr Ali Khairbek (Post Doc)
- Dr Zakir Ullah (collaborator)
Computational catalysis has emerged as a cornerstone of modern chemical research, offering an indispensable microscope to observe the transient states and electronic rearrangements that define catalytic efficiency. By using high-level quantum mechanical calculations, we can move beyond trial-and-error experimentation in the coventional lab to rationally design catalysts that address pressing global challenges in green energy and sustainable synthesis. The future of this field lies in the development of autonomous, AI-enhanced protocols and quantum-mechanical workflows that can predict reaction pathways with unprecedented accuracy, transforming how we develop the next generation of industrial and environmental materials.
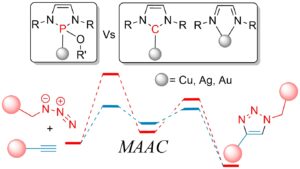
We also do the high-level computational exploration of catalytic mechanisms, particularly 32CA and ethylene transformations. Extensive DFT investigations using functionals like MN12-L and MO6-2X have elucidated the reactivity of novel RhCp-X complexes, where RhCpSO3H emerged as a premier catalyst for 1,5-triazole synthesis due to enhanced stability and lower activation barriers. In the realm of RuAAC, halogen substitution on Cp RuX catalysts was found to significantly modulate catalytic performance, with the 1,5-pathway remaining the most energetically favorable across F, Cl, Br, and I variants. The assessment of ADAP-M and NHC-X catalysts revealed that binuclear pathways often provide superior transition state stabilization compared to mononuclear routes, with Cu-based systems and Cl-substituted NHC ligands exhibiting the highest efficiency in synthesizing 1,2,3-triazoles.
Investigations into organocatalysis and metal-free synthetic routes emphasize sustainable chemical development. The group identified pyrrolidine as a highly effective amine catalyst for the regioselective synthesis of N-arylbenzotriazoles, while studies on the 32CA of azides with enaminones highlighted the role of water in lowering gree energy and promoting the 1,4-pathway through solvent-assisted tautomerization. These studies are complemented by mechanistic analyses of Ni(II)-catalyzed ethylene oligomerization via the Cossee-Arlman mechanism, where NON-type ligands with Ph substituents were shown to effectively suppress beta-H elimination and favor trimerization by lowering energy barriers for the dimerization and trimerization processes.
Analytical depth is provided by advanced computational tools including NCI and RDG to visualize weak interactions and ELF and LOL to assess covalent stability. By utilizing FMO and ADCH charge analyses, the group precisely maps regioselectivity and electronic properties, ensuring a robust theoretical foundation for the design of novel catalysts. These methodologies facilitate the detailed assessment of steric hindrance and electronic stabilization, driving the discovery of efficient catalysts like NHSi- and NHGe-supported CuBr for CuAAC reactions. This comprehensive approach enables the group to resolve complex molecular recognition patterns and optimize the performance of both transition-metal and metal-free catalytic systems.
Chemical Bond – Sulphur centered H Bond, Halogen bond & Tetral bond
- Sneha Anna Sunny (PhD student)
- Alen Binu Abrahan (Visiting researcher from Austonomous Universityof Madrid, Spain and Erasmus Mundus Fellow)
- Arnav Paul (NIISER , now PhD Student at University of Illinois at Urbana-Champaign)
- Mebin Varghese (Visitor, now PhD Student at VIT)
- Dr Aristote Matando (co-supervised PhD student, University of Kingshasa, Congo)
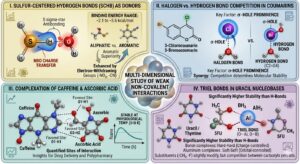
A. Paul, R. Thomas, Evidences for sulfur centered hydrogen bond with sulfur atoms as a donor in aromatic thiols and aliphatic thiols in aqueous solution, J. Mol. Liq. 348 (2022). https://doi.org/10.1016/j.molliq.2021.118078.
Our primary contribution to the field of molecular science lies in the definitive characterization of sulfur-centered hydrogen bonds (SCHB) where the sulfur atom unexpectedly acts as the hydrogen bond donor. While conventional chemical wisdom often positions sulfur as an acceptor due to its lower electronegativity compared to oxygen, our research established that in both aliphatic and aromatic thiols, the sulfur atom forms significant S-H…. O interactions with water molecules. We discovered that aromatic thiols possess a superior capacity for these interactions compared to aliphatic ones, and we further demonstrated that the introduction of electron-withdrawing groups, such as nitro or cyano groups, substantially enhances the hydrogen bond strength. By quantifying these interactions, we categorized them as weak non-covalent bonds with specific binding energies typically ranging from -2.1 to -3.6 kcal/mol, providing a new benchmark for understanding sulfur’s role in aqueous environments.
M. Varghese, J.M. Thomas, A.Y. Alzahrani, R. Thomas, Exploring the dynamics of halogen and hydrogen bonds in halogenated coumarins, Zeitschrift Fur Phys. Chemie. (2024). https://doi.org/10.1515/zpch-2023-0570.
A.B. Abraham, A.Y. Alzahrani, R. Thomas, Exploring non-covalent interactions between caffeine and ascorbic acid: their significance in the physical chemistry of drug efficacy, Zeitschrift Fur Phys. Chemie. 238 (2024) 401–420. https://doi.org/10.1515/zpch-2023-0390.
[152] G. Ashok, F. Thomas, R. Thomas, Understanding the hydrogen bonding preferences and dynamics of prontosil in water and methanol, Chem. Phys. Impact. 8 (2024).
Beyond sulfur chemistry, we have made significant strides in mapping the competitive landscape of weak bonds, particularly the interplay between halogen and hydrogen bonding in biologically active coumarins. Our work revealed a sophisticated competition in molecular stabilization where hydrogen bonds generally dominate; however, we proved that halogen bonds involving bromine and iodine are crucial contributors to stability due to the presence of a significant sigma-hole. We also quantified the molecular-level interactions of common substances like caffeine and ascorbic acid, identifying the specific favorable sites that facilitate their complexation and influence drug efficacy. By establishing that these weak-bonded complexes remain thermally stable at physiological temperatures (310 K), our research provides a vital foundation for predicting how pharmaceutical compounds and biomolecules behave within the human body.
A. Matondo, R. Thomas, P.V. Tsalu, C.T. Mukeba, V. Mudogo, -methylation and -fluorination electronic effects on the regioselectivity of carbonyl groups of uracil by H and triel bonds in the interaction of U, T and 5FU with {HCl} and {TrH}3 (Tr~= B, Al), J. Mol. Graph. Model. 88 (2019) 237–246. https://doi.org/10.1016/j.jmgm.2019.02.006.
Our research into non-covalent interactions addresses the fundamental challenge of understanding how substituents tune the regioselectivity and bioactivity of uracil-based drugs, which is critical for optimizing their performance in pharmaceutical and anticancer applications. By investigating the electronic effects of alpha-methylation and alpha-fluorination, we have contributed to the relatively sparse literature on triel-bonded complexes, specifically identifying that these interactions O….. Al and O……B are significantly more stable than traditional hydrogen bonds. Our findings reveal that while the triel atom’s size influences the nature of the bond shifting from a charge-controlled hard-hard interaction in boron to an orbital-controlled soft-soft interaction in aluminum—the overall impact of substituents on bond strength remains remarkably small. This leads to the significant conclusion that a persistent competition exists between the two carbonyl groups of the uracil skeleton, a discovery that underscores the complexity of molecular recognition and provides a vital framework for predicting the behavior of nucleobase derivatives in biochemical processes.
Weak Interactions, Microsolvation and its dynamics
- Sneha Anna Sunny (PhD student)
- Francis Thomas (PhD student)
- Dr. T. Pooventhiran (post doc, Currently at IISER and KR College)
- Dr Jisha Mary Thomas (Post Doc, currently at CHRIST)
The field of theoretical chemistry has undergone a significant paradigm shift with the ability to precisely model non-covalent interactions (NCIs). Despite their relative weakness compared to traditional covalent bonds, NCIs are the fundamental organizing principles that dictate molecular recognition, protein folding, and supramolecular assembly. Our research focuses on deconstructing these subtle, dispersion-driven forces using high-level computational modeling and quantum chemistry calculations. This approach is not merely a theoretical exercise; it is an essential tool for unravelling complex reaction mechanisms and advancing the design of more efficient, environmentally friendly chemical processes. Through our recent investigations, we have achieved significant milestones in understanding the solvation dynamics of bioactive molecules and materials. We have successfully characterized the specific hydrogen-bonding motifs between caffeine and ascorbic acid, shedding light on the molecular-level interactions that influence drug efficacy and delivery. We have also ystematically analysed the microsolvation of amino acids and various drug molecules.
R. Thomas, T. Pooventhiran, Study of the dynamics of the interaction of glycine and GABA with water and ethanol using theoretical tools, J. Mol. Liq. 368 (2022). https://doi.org/10.1016/j.molliq.2022.120721.
R. Thomas, T. Pooventhiran, M.A. Bakht, A.Y. Alzahrani, M.A. Salem, Study of interaction between different solvents and neurotransmitters dopamine, l-adrenaline, and l-noradrenaline using LED, QTAIM and AIMD, J. Mol. Liq. 368 (2022). https://doi.org/10.1016/j.molliq.2022.120708
Synthesis of bioactive compounds
- Rajimon KJ
- Dr N Elangovan (Post Doc)
Earlier, our group synthesised and investigatigated the biological activities of Schiff bases and chalcones, recognizing their importance in both synthetic chemistry and pharmacology. Schiff bases, formed through the condensation of primary amines and carbonyl compounds, are versatile intermediates known for their role in organic synthesis, particularly in drug development, owing to their diverse biological activities, including anticancer and antimicrobial properties. Chalcones, α,β-unsaturated ketones, serve as essential precursors in the synthesis of various biologically active compounds, such as flavonoids, and possess pharmacological significance, with antioxidant, anti-inflammatory, and anticancer attributes. Our group is committed to designing, synthesizing, and evaluating novel Schiff bases and chalcones, with a focus on understanding their interactions with biological systems, including enzymes and receptors. Dr Rajimon KJ’s PhD was in this area.
K.J. Rajimon, N. Elangovan, A. Amir Khairbek, R. Thomas, Schiff bases from chlorine substituted anilines and salicylaldehyde: Synthesis, characterization, fluorescence, thermal features, biological studies and electronic structure investigations, J. Mol. Liq. 370 (2023). https://doi.org/10.1016/j.molliq.2022.121055.
P. Surendar, T. Pooventhiran, N. Al-Zaqri, S. Rajam, D. Jagadeeswara Rao, R. Thomas, Synthesis of three quasi liquid Schiff bases between hexanal and adenine, cytosine, and l-leucine, structural interpretation, quantum mechanical studies and biological activity prediction, J. Mol. Liq. (2021) 117305. https://doi.org/https://doi.org/10.1016/j.molliq.2021.117305.
Get In Touch
We welcome research collaborations and enquiries from theoreticians, experimental chemists, and interdisciplinary researchers. Prospective PhD, postdoctoral, and student researchers are encouraged to contact us via the details below or the message form.
Dr. Renjith Thomas
Department of Chemistry
St. Berchmans College (Autonomous)
Changanassery, Kerala, India – 686101
[email protected]
+91 95446 58314